
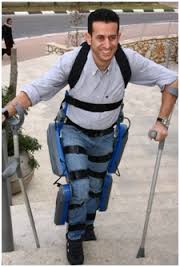
Photograph of the ReWalk courtesy of Argo Medical Technologies.
On June 26, the U.S. Food and Drug Administration (FDA) announced that it will allow marketing of the ReWalk Personal System robotic exoskeleton, developed by Argo Medical Technologies, Yokneam Illit, Israel. The company’s U.S. office operates as ReWalk Robotics, Marlborough, Massachusetts. The ReWalk Personal is designed for home and community use by people with paraplegia caused by spinal cord injury at levels T7 (seventh thoracic vertebra) to L5 (fifth lumbar vertebra). The device allows the user to sit, stand, and walk with assistance from a trained caregiver. Patients and their caregivers must undergo training developed by the manufacturer to learn and demonstrate proper use of the device.
The FDA reviewed the ReWalk Personal through its de novo classification process, a regulatory pathway for novel medical devices that are generally low to moderate risk. The FDA is requiring Argo Medical Technologies to complete a post-market clinical study that will consist of a registry to collect data on adverse events related to the use of the ReWalk Personal and prospectively and systematically assess the adequacy of its training program.
To assess the safety and effectiveness, the FDA reviewed testing that assessed the device’s durability, its hardware, software, and battery systems, and other safety systems that help minimize risk of injury should the device lose balance or power. The FDA also reviewed clinical data based on 30 study participants. The clinical tests assessed the participants’ ability to walk various distances, the amount of time needed to walk various distances, performance on various walking surfaces and slight slopes, and performance walking in areas where jostling might occur. Studies also assessed the risk of certain physical effects on the user. Additionally, observational data from 16 patients were provided to support use of the device on various walking surfaces in the home and community with various levels of assistance from a trained companion.


